The MDCG requires 8 steps

The coordination group of medical devices (MDCG) has designed an orientation document that describes how their producers Medical devices in class 1 Complicators mdr.
The document is entitled Orientation notes for producers of class I medical devices. This article summarizes this document and offers manufacturers of these products.
1. What does medical devices distinguish in class 1
a) Classification of medical devices in classes
The order of the MDR medical device divides medical devices into classes 1, 2nd, 2b and 3 .. the same goes for the guideline of the MDD medical device. Lessons are often written also with Roman figures (class I, IIA, IIB and III).
The classification rules assign higher risks to higher classes.
Examples of class I medical devices and many surgical tools such as tweezers, latex gloves, masks, wheelchairs (without motor), stethoscopes, associations or glasses.
b) effect of classification
The classes do not have (!) Influence on the basic security and performance requirements, whose manufacturers must demonstrate. Rather, classes determine the conformity assessment procedure in order to demonstrate compliance with these safety and performance requirements.
For class 1 medical devices, points appointed in the conformity evaluation procedure is not included. In addition, the MDR does not insist on the certification of the quality management system by an area called in class 1 medical devices. It is saving time and costs.
An exception are medical devices in the 1S, 1r and 1m classes:
- 1S: products that are positioned on the market in sterile conditions
- 1r: reusable surgical instruments (r is for “reusable”)
- 1m: Products with measurement function
For these “1*products, producers must integrate the positions called in the assessment of compliance.
2. How class 1 medical devices are approved
The MDCG describes eight steps that producers should pass on the market.
Step 1: review and confirm that the product is a medical device
The MDR adopted the definition of the term “medical device” almost unchanged by the MDD. It is therefore unlikely that a product that was previously a medical device does not go down below the scope of the MDR.
Step 2: Check that it is a class 1 medical device
This step is essential because the MDR has changed the classification rules. For example, almost any autonomous software now falls Not More in this lower class!
The MDCG orientation document, of which this article discusses, can only be used for class 1 medical devices, even if most of the requirements apply to all medical devices.
Step 3.a: make sure that the basic security and performance requirements are satisfied
It is obvious that the basic security requirements and performance of the appendix I must be satisfied. However, the MDCG stresses that other regulations such as the machine directive can also be observed.
It underlines the importance of risk management and recalls that the test of the requirements is possible by using harmonized rules and common specifics.
Step 3.B: Create a clinical evaluation
The MDCG underlines how important the MDR is the clinical evaluation and that the MDR insists that the producers
Step 3.c: Create technical documentation
Producers of class 1 medical devices must create technical documentation in accordance with attachments II and III. Of course, this applies to all medical devices.
The MDCG document responds to some changes introduced by the MDR compared to the MDD:
- Reason for the classification
- Bissis Udi-di
- Reference to predecessors and similar products
- Reference to harmonized rules applied and therefore valid and “specific common”
- Description of the Post-Market-Surveillance system
Step 3.d: Request when named
As already mentioned above, class 1* producers must include an accredited notified area for the classes of corresponding products:
- Devices in sterile conditions: MDS 1005 code
- Reusable surgical tools: MDS 1006 code
- Devices with a measurement function: MDS 1010 code
Manufacturers can see in the Nando database which are credited to the respective code.
Step 3.e: Prepare the instructions for use and labeling
It is somehow surprising that only to passage 3. and the MDCG faces the instructions for use and labeling. Finally, the appendix I determines its content. Clinical evaluation also requires instructions for use.
The MDCG mentions that for medical devices in class I there are no instructions for use if the safe use is guaranteed. The document also enters the languages (without referring to a list of required languages) and the duty of retailers to provide the accompanying materials in these languages.
The symbols are specified through harmonized rules and common specifics; The label must clarify that the product is a medical device.
Step 4: check compliance with the requirements for manufacturers
This fourth step concerns less the product than the manufacturer, it goes for its obligation to establish a QM system and eliminate insurance.
The MDCG does not respond to the new obligation according to a person responsible in accordance with article 15, but already in the introduction.
Step 5: Create declaration of conformity
It is not surprising that the requirement that the producers declare compliance with the MDR and, if necessary, further EU regulations and that this declaration of conformity must translate into national languages in which the product is offered.
Step 6: Apply the EC marking
This step is also obvious: the producers of class I medical devices must also attach the EC marking, so the number of the area called must be part of this label for class 1 products.
Step 7: Product registration and manufacturer in Eudamed
The seventh step is new in this form: the producers are required to register in the Eudamed, after which a “srn” is assigned. Incidentally, the same goes for EU representatives and importers.
So it is the task of producers to record medical devices. Udi-di and the Udi-di base are assigned.
Until Eudamed is fully functional, the EU representatives or representatives must inform the responsible authority (in Germany the BFARM) or record the product.
Step 8.a: collect and evaluate the post-market data
The MDR obliges manufacturers to establish a PMS system that is part of the QM system. For class 1 medical devices, producers must create a monitoring report after positioning on the market, the post-market surveillance report (PMS-Report =. The requirements for this report are lower than the periodic security update reports (PSUR).
Step 8.B: Vigilanz system
The MDCG recalls that the MDR also obliges the producers of class 1 products, Corrective actions for field safety (Fsca). Until the Eudamed is not in operation, these relationships go to the authorities (in Germany BFARM, Swiss SwissMedic in Switzerland). These reports are subsequently recorded in Eudamed.
The MDCG document describes the duties in the case of Fsca in a relatively large way, as is currently (still) the order of the safety plan of medical devices.
Step 8.c: it is non -compliant products
The last step concerns how to manage non -compliant products. In addition to the reporting obligations already mentioned (phase 8.b), manufacturers must adopt appropriate corrections or corrective measures.

The fastest and cheap way to allow your class I product
With our video and dozen models training you will receive everything you need for quick and compliant with MDR/IVDR. Individual onboarding and a tailor -made project plan for you make you easy to maintain an overview and pass through all the necessary steps.
3. Which authorities control class I
Contrary to higher class products, the monitoring of class I and their producers are borne by the authorities. These authorities, e.g. B. Commercial authorities and regional councils, this monitoring obligation meet more and more. They are based on control lists as the following exemplary list. However, please note that not all control lists seem the same.
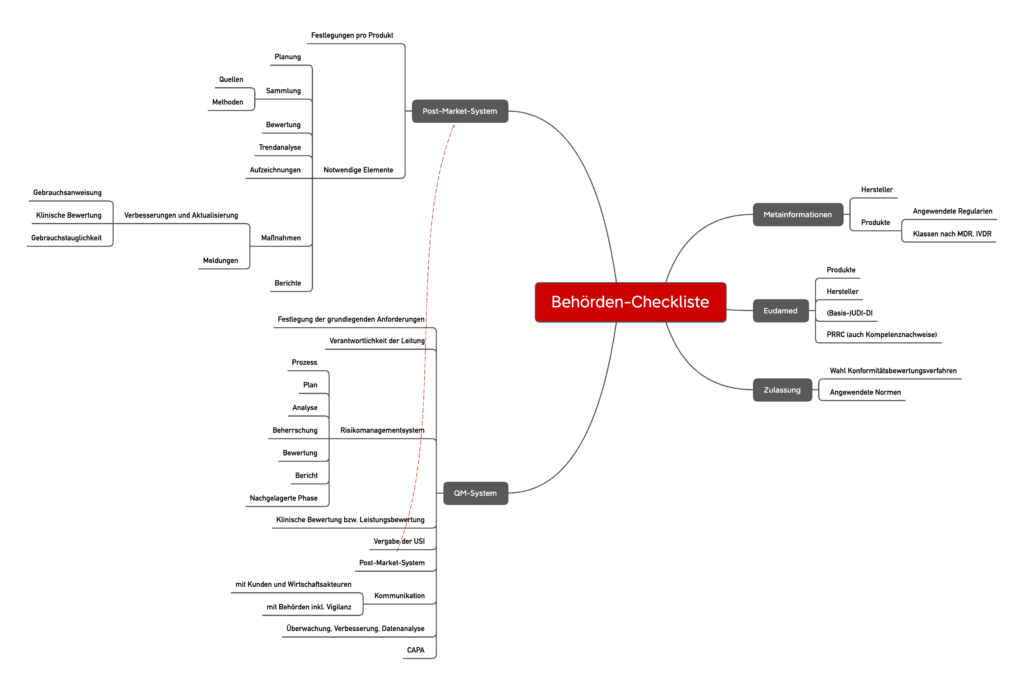
As expected, the control lists of the authorities are strongly based on the MDR or the IVDR. The focus of this control list is surprising on the specifications for the risk management system, post-market surveillance and vigilance. Here the authorities respond to almost every part of the regulatory requirements.
4. Conclusion
The 20 pages document Orientation notes for producers of class I medical devices It should – as the name suggests – serve as producers of guides of class I medical devices.
The document does justice to this goal. On the other hand, it must be clear that the requirements mentioned there are not completely and essentially non -specific for class 1 medical devices.
The Johner Institut supports class I manufacturers in meeting compliance with regulatory requirements quickly and simple and therefore be perfectly prepared for the inspections of the authorities. Come into contact with us, e.g. B. through our web module.
Change the story
- 2025-03-28: examples in section 1. A) inserted
- 2023-03-04: Notes on the articles on class I software I integrated
- 2021-09-26: numbered header and chapter 3 inserted.
Similar posts
The coordination group of medical devices (MDCG) has designed an orientation document that describes how their producers Medical devices in class 1 Complicators mdr. The document is entitled Orientation notes for producers of class I medical devices. This article summarizes this document and offers manufacturers of these products. 1. What does medical devices distinguish in…