Biokompatibilität ISO 10993 – Materialzertifikate reichen nicht aus!

Regularien wie die MDR fordern den Nachweis der Biokompatibilität aller Materialien, die mit Patienten oder Anwendern direkt oder indirekt in Kontakt kommen.
Eine prüfbasierte Beurteilung der Biokompatibilität liefert Referenzdaten, die bei unerwarteten Problemen ein sehr gutes Fundament liefern und helfen, unbekannte Ursachen sehr schnell und gezielt zu identifizieren.
Allerdings gibt es seit Jahren einen gegensätzlichen Trend. Weg von Prüfungen und damit weg von der Produktsicherheit.
1. Grundlagen
a) Risiken durch mangelnde Biokompatibilität
Medizinproduktehersteller sind verpflichtet, die Sicherheit ihrer Produkte zu gewährleisten und mögliche Risiken zu minimieren. Zu diesen Risiken zählen auch biologische Risiken, die beispielsweise verursacht werden durch:
- Chemikalien, die sich aus Materialien des Medizinprodukts herauslösen und vom Körper aufgenommen werden
- Implantate, die eine Abstoßungsreaktion hervorrufen
- Materialien, die eine allergische Reaktion verursachen
- Materialien, die entgegen der Absicht des Hersteller vom Körper abgebaut oder umgewandelt werden
- Implantate und Materialien, die nicht wie gewünscht in den Körper einwachsen
Faktoren, die die Biokompatibilität negativ beeinflussen, beschreibt dieser Artikel weiter unten.
b) Begriffsdefinitionen
Die ISO 10993 definiert den Begriff biologisches Risiko wie folgt:
„Wahrscheinlichkeit von gesundheitlichen Schäden aufgrund des Medizinprodukts oder Wechselwirkungen mit den Materialien.“
Quelle: ISO 10993-1
Die biologische Sicherheit ist entsprechend die Freiheit von nicht akzeptablen biologischen Risiken.
Etwas kryptischer wirkt die Definition des Begriffs „Biokompatibilität“:
„Fähigkeit eines Medizinprodukts oder Materials, mit einer angemessenen Host-Reaktion Leistung in einer spezifischen Anwendung zu erbringen“
Quelle: ISO 10993-1
Der Begriff Host-Reaktion meint hier mögliche unerwünschte und nachteilige Reaktionen. Die Norm merkt an, dass der Nachweis der Biokompatibilität durch biologische Prüfungen und durch eine Beurteilung der herauslösbaren Chemikalien erfolgen kann.
2. Regulatorische Anforderungen an die Biokompatibilität
a) Anforderungen der MDR
Die Medizinprodukteverordnung (Medical Device Regulation MDR) räumt der Biokompatibilität einen hohen Stellenwert ein, wie man bereits in den Erwägungsgründen erkennt. Sie formuliert die entsprechenden Anforderungen an mehreren Stellen:
Anhang I: Grundlegende Sicherheits- und Leistungsanforderungen
Der Anhang I erwähnt den Begriff Biokompatibilität nicht explizit. Er schreibt aber:
Die Produkte werden so ausgelegt, hergestellt und verpackt, dass die Risiken durch Schadstoffe und Rückstände für Patienten — unter Berücksichtigung der Zweckbestimmung des Produkts — sowie für Transport-, Lager- und Bedienungspersonal so gering wie möglich gehalten werden. Dabei wird Geweben, die diesen Schadstoffen und Rückständen ausgesetzt sind, sowie der Dauer und Häufigkeit der Exposition besondere Aufmerksamkeit gewidmet.
MDR, Anhang I, Absatz 10.2
Bereits im ersten Abschnitt des Anhangs I fordert die Verordnung, die Sicherheit und Gesundheit von Patienten, Anwendern und Dritten nicht zu gefährden und die „Risiken durch sichere Auslegung und Herstellung beseitigen oder so weit wie möglich [zu] minimieren“.
Anhang II: Technische Dokumentation
Die MDR verpflichtet die Hersteller im Anhang II, Informationen zu allen Prüfungen zu dokumentieren inklusive
Testaufbau, Testprotokolle, Methoden der Datenanalyse, […], Testergebnisse hinsichtlich der Biokompatibilität des Produkts einschließlich der Identifizierung aller Materialien in direktem oder indirektem Kontakt mit dem Patienten oder Anwender, [sowie hinsichtlich der …] chemischen und mikrobiologischen Parameter.
MDR, Anhang II, Absatz 6.1.b)
Auch den Fall, dass die Hersteller keine Tests durchführen, regelt die MDR:
Falls keine neuen Tests durchgeführt wurden, wird diese Entscheidung in der Dokumentation begründet. Eine solche Begründung wäre beispielsweise, dass Biokompatibilitätstests an identischen Materialien durchgeführt wurden, als diese Materialien in ein rechtmäßig in Verkehr gebrachtes oder in Betrieb genommenes Vorgängermodell des Produkts integriert wurden.
MDR, Anhang II, Absatz 6.1.b)
Anhang VII: Anforderungen an Benannte Stellen
Dieser Anhang betrifft die Hersteller zumindest indirekt, denn darin verpflichtet die MDR die Benannten Stellen:
- Die Kompetenz des Personals der Benannten Stellen bezüglich der Biokompatibilität gewährleisten
- Aktualität des Wissens der Hersteller
Benannte Stellen müssen die Verfahren der Hersteller prüfen, mit denen die Hersteller sicherstellen, dass sie über neuste wissenschaftliche Erkenntnisse auch in Bezug auf die Biokompatibilität von Materialien verfügen.
b) ISO 10993-1
Die Norm ISO 10993-1 war bereits unter der Medizinprodukterichtlinie MDD harmonisiert und wird das (wie die ganze Normenfamilie) auch unter der MDR bleiben.
Daher orientieren sich die meisten Medizinproduktehersteller an dieser Norm, u.a. wenn es um Endpunkte geht, die für Bewertung der biologischen Sicherheit notwendig sind.
Diese Endpunkte beschreibt die Norm in Tabelle A.1 im Annex A. Der folgende Ausschnitt aus der Tabelle zeigt exemplarisch die zu bewertenden Endpunkte für ein Medizinprodukt mit Kontakt zu zirkulierendem Blut.
Diese im nicht-normativen Teil beschriebene Vorgehensweise entspricht nicht den normativen Vorgaben: Gemäß ISO 10993-1 (2018), Figure 1, sowie ISO 10993-2 ist ausdrücklich eine Materialcharakterisierung durchzuführen und auf Tierversuche zu verzichten. Zudem ermöglicht eine Materialcharakterisierung eine höhere Aussagekraft und damit eine höhere biologische Sicherheit.
Die Norm stellt klar, dass die Hersteller die biologische Sicherheit über den kompletten Lebenszyklus des Medizinprodukts gewährleisten müssen:
„The biological safety of a medical device shall be evaluated by the manufacturer over the whole life cycle of a medical device.“
ISO 10993-1, 4.7
Ebenso ergänzt dieser Teil die Forderungen nach „Freiheit von inakzeptablen biologischen Risiken“ wie folgt:
„The following shall be taken into account for their relevance to overall biological evaluation of the medical device: …process contaminants and residues… packaging materials… leachables substances… degradation products…“
ISO 10993-1, 4.3
Bitte beachten Sie auch diese Fachartikel:
c) Anforderungen der FDA
Die FDA bietet ein ausführliches Guidance-Dokument zur ISO 10993-1 für die Bewertung der Biokompatibilität an, welches sowohl das Vorgehen zur Bewertung der Biokompatibilität nach ISO 10993-1 im Detail beschreibt als auch zusätzliche Anforderungen wie weitere Endpunkte für bestimmte Produktkategorien angibt. Die fehlenden Endpunkte in der ISO 10993-1 sind ein Grund, warum die ISO 10993-1:2018 nur teilweise von der FDA anerkannt wird.
In der 2023 veröffentlichten Version des Dokuments wurde der Anhang G „Biocompatibility of Certain Devices in Contact with Intact Skin“ hinzugefügt, um den „3R-Ansatz“ zu Reduzierung, Verbesserung und Ersatz von Tierversuchen weiter zu unterstützen.
Im September 2024 hat die FDA zudem einen neuen Entwurf eines Guidance-Dokuments zur Biokompatibilität veröffentlicht, das sich speziell mit der Durchführung chemischer Analysen befasst.
- Ziel
Ziel des Dokuments ist die Standardisierung der Vorgehensweise. Es schließt damit den Spielraum, den die Norm lässt. - Fokus
Der Fokus der Guidance liegt auf Extractables-Studien, also der Identifizierung und Quantifizierung von Substanzen, die nach der Extraktion des Medizinprodukts in Lösungsmitteln freigesetzt werden. - Inhalt
Die Guidance bietet detaillierte Empfehlungen zur Durchführung der Extraktion, zur chemischen Analytik sowie zur Notwendigkeit einer umfassenden und präzisen Dokumentation – ergänzend zur ISO 10993-18. - Zielgruppe
Das Dokument wendet sich v. a. an Toxikologen, Biokompatibilitätsbewerter und Experts für toxikologische Risikobewertungen, aber vor allem auch Labore, die chemische Analysen durchführen. - Kritik
Kritisch anzumerken ist, dass die Anforderungen an den Umfang der chemischen Analysen, die Datenqualität und insbesondere die Dokumentation sehr hoch sind. Dies dient der Erhöhung der Produktsicherheit, könnte jedoch in einem unverhältnismäßigen Anstieg des Aufwands und der Kosten für Laborprüfungen resultieren – selbst bei bewährten Standardmaterialien.
Fazit: Die Anforderungen an die Biokompatibilität steigen. Daher ist es wichtig, die Laboruntersuchungen sehr gezielt auszuwählen und durchzuführen.
Wir unterstützen Sie bei der vollständigen Planung und Bewertung der Biokompatibilität Ihrer Produkte. Wir übernehmen die Organisation der Laborprüfungen und die Erstellung der erforderlichen Dokumentation. Dank unserer Erfahrung können wir die Prüfparameter auf das jeweilige Produkt und Material gezielt abstimmen. Dadurch sparen Sie Kosten und erreichen dennoch Konformität mit den Anforderungen der FDA.
3. Umgang mit Informationen zur Biokompatibilität
a) Vorhandene Informationen nutzen
Zur Beurteilung der Biokompatibilität tragen Hersteller häufig alle Informationen zu den verwendeten Materialien zusammen wie
- Materialdatenblätter
- Zertifikate
- Ergebnisse von Recherchen zu bekannten Inhaltsstoffen und Additiven
- Markterfahrungen
Diese Informationen sind notwendig und wichtig, aber meist nicht hinreichend, um die Biokompatibilität zu beweisen!
b) Fehlende Informationen ergänzen
Die oben genannten Informationen sind eine sehr gute Ausgangsbasis. Häufig erlauben sie aber noch keine vollständigen Aussagen über die biologische Sicherheit. So fehlen Informationen, die nachweisen, dass es keine biologischen Risiken gibt durch die
- Wechselwirkungen der verschiedenen Materialien untereinander
- Veränderungen der Materialien über die Lebensdauer des Produkts
- Bearbeitung der Materialien im Rahmen der Produktion
- Endreinigung des Produkts
- usw.
Die Liste möglicher Einflussfaktoren ist sehr umfasend: Sie reicht vom Handdesinfektionsmittel der Mitarbeiter, über unreine Ausgangsmaterialien, bis hin zu nicht wahrgenommenen Kabelabrieb an den Anlagen. Alle Faktoren der folgenden Tag-Cloud haben bereits erfolgreich eine positive Bewertung der Biokompatibilität verhindert.

c) Vorhandene Informationen kritisch bewerten
Auch die Verlässlichkeit der vorhandenen Informationen sollten Hersteller kritisch hinterfragen und beispielsweise folgende Aspekte klären:
- Prüfverfahren und Prüfparameter
- Ausgangsmaterialen, mit denen diese Ergebnisse gewonnen wurden
- Übertragbarkeit der Ergebnisse
Dass Hersteller auf Informationen von Mitbewerbern zurückgreifen, ist gut. Jedoch sollten Sie bei der Argumentation, dass deren Produkte ausreichend ähnlich und seit Jahren ohne Probleme auf dem Markt verwendet würden, die Äquivalenz aller Parameter betrachten wie:
- Sämtliche Inhaltsstoffe und Materialtypen wie Additive und Einfärbungen und nicht nur die Hauptbestandteile
- Produktionsprozess und Produktionshilfsmittel
- Endreinigung: Prozess, Reinigungsmittel
Häufig fehlen genau diese Informationen und führen zu argumentativen Lücken. Im Datenblatt nicht gelistete Inhaltsstoffe sind nicht automatisch unkritisch.
4. Weitere Praxistipps
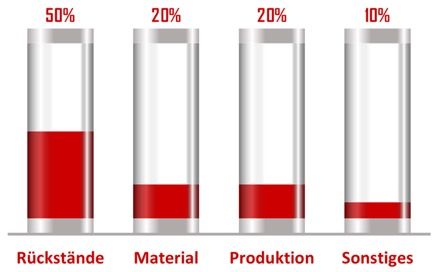
Die folgende Grafik spiegelt die Erfahrung des Johner Instituts mit den typischen Problemen bei mehreren 1000 Medizinprodukten wider.
a) Nicht auf „etablierte Meinungen“ verlassen
Manche Hersteller folgen der Ansicht, dass der Cytotoxizitätstest alleine ausreichend würden, um die biologische Sicherheit zu gewährleisten, oder dass Produkte mit sehr kurzem Haut- oder Schleimhautkontakt als unkritisch anzusehen sind. Beides ist nicht zutreffend und nicht konform mit den o.g. Anforderungen der MDR.
b) Notwendige Nachweise bereitstellen
Leider verwirren sowohl die ISO 10993 als auch die MDR durch mehrere Aussagen, welche bei einigen Produkten den Rückschluss ermöglichen, dass keine Prüfungen benötigt würden. An anderen Stellen finden sich gegenteilige Behauptungen. Daher zwei konkrete Praxistipps:
- Was die Benannten Stellen sehen wollen
Fast immer wollen die benannten Stellen Prüfungen zu genau den in der Tabelle genannten Endpunkten sehen. Oder zumindest eine Begründung, warum diese weggelassen/ersetzt wurden. - Effekte, die Hersteller diskutieren müssen
Es ist meist nicht möglich, Effekte apriori auszuschließen, die sich ergeben durch Reinigungsmittelrückstände, Produktionsrückstände, Sterilisation, die Aushärtung eines Klebers, das Abdampfen von Lösungsmitteln, Effekte durch Gravuren, Bedruckungen, Materialverunreinigungen beim Extrudieren, unzureichende Polymerisation, Verpackungskomponenten, unzureichende Materialqualitäten, unzulässige Weichmacher oder andere Additive, durch Einfärbungen usw.
c) Keine minderwertigen Materialien verwenden
Mit hochwertigen Ausgangsmaterialien lassen sich viele Probleme von Beginn an vermeiden. So sind zum Beispiel bei einem minderwertigen Polyurethan (PUR) weit über 200 verschiedene Extractables möglich, wenn auch nicht alle auf einmal. Bei einem sehr guten PUR sind hingegen keine Leachables zu erwarten.
d) Prüfungen: Risikobasiert arbeiten
Es benötigt viel Erfahrung, um eine Grenze zu setzen, ab der ein Material nicht mehr als akzeptabel bewertet werden kann. Und natürlich sind diesbezüglich umfangreiche Kenntnisse zu Grenzwerten unerlässlich.
Die meisten Medizinprodukte sind bei der Prüfung der Biokompatibilität unauffällig. Das Verhältnis hängt stark vom Material und Produkttyp ab. Bei den auffälligen Produkten können aber 90% der Abweichungen direkt oder mit ergänzenden Analysen als akzeptabel argumentiert oder durch eine unkomplizierte Optimierung behoben werden.
Nutzen Sie dabei Erfahrungen, um Risiken besser einschätzen zu können, wie:
- Produkt und Produkttyp (Historie)
- Ähnliche Produkte
- Materialien
- Aufbereitung und Produktion des Produkts
5. Fazit
Der Nachweis der Biokompatibilität lässt sich nicht ausschließlich anhand von Datenblättern führen. Daher sind Prüfungen notwendig.
Allerdings sollten diese Prüfungen nicht abschrecken, weil die Kosten dafür sehr überschaubar sind. Das ist die Erfahrung der Experten für Biokompatibilität beim Johner Institut, die bereits mehrere 1000 Medizinprodukte getestet haben.
Diese Minimierung des Prüfungaufwands ist möglich, falls man die richtige Strategie wählt und
- vorhandene Informationen sorgfältig recherchiert,
- mögliche Einflussfaktoren auf die Biokompatibilität zusammenträgt,
- aus beidem Rückschlüsse ableitet und Beweisketten erstellt und
- durch einen risikobasierten Ansatz die Aufwände für die Prüfungen minimiert z. B. unnötige Prüfungen vermeidet.
Änderungshistorie
- 2024-09-27: Kapitel 2.c) eingefügt
- 2024-05-02: Hinweise auf weiterführende Artikel ergänzt. Redaktionelle Verbesserungen
- 2019-07-22: erste Version des Artikels erstellt
Ähnliche Beiträge
Regularien wie die MDR fordern den Nachweis der Biokompatibilität aller Materialien, die mit Patienten oder Anwendern direkt oder indirekt in Kontakt kommen. Eine prüfbasierte Beurteilung der Biokompatibilität liefert Referenzdaten, die bei unerwarteten Problemen ein sehr gutes Fundament liefern und helfen, unbekannte Ursachen sehr schnell und gezielt zu identifizieren. Allerdings gibt es seit Jahren einen gegensätzlichen…