Tipps Zum FDA Guide “Interoperable medical devices”

Die FDA The importance of medical devices’ abuse recognized at the beginning and 2017 Dokument Guida ‚Interoperable medical devices’ Published.
The American authority would like to take into account the fact that the interoperability of medical devices is important for health care on the one hand. On the other hand, problems with a lack of interoperability lead to risks.
This article gives you a quick overview of the FDA requirements to “interoperable medical devices” and gives advice on how to meet them.
1. Information on the orientation document
A. If the FDA document concerns
The orientation document “Interoperable medical devices” is addressed ProducerYou want to sell medical devices with data interfaces in the United States. The authority formulates its expectations towards “admission documents”.
In addition, FDAMIT also wants your document at the same time Inspectors transmit the understanding of their authority on this topic.
Like all “orientation documents”, this is not “legally binding”. However, the FDA provides that manufacturers contain the information requested in this document for presentations (for example all forms of 510 (K), PMA)!
In addition, the producers that import in the United States are generally well recommended to follow all the “orientation documents” of the FDA.
At the same time, this document also provides producers of medical devices outside the US market precious information on the development of interoperable medical devices.
B. What the FDA includes for interoperability
The FDA follows the usual definitions of the term “interoperability”. Consequentially The ability of two or more products, technologies or systems, to exchange information and use information exchanged.
In this article, read more about what you mean by interoperability and what levels of interoperability differ.
2. Requirements for “interoperable medical devices”
The document, which is pleasantly short with 18 pages, introduces the topic into the first four chapters and appoints the area of application and definitions.
Essential needs can be found in chapters V and VI:
- Chapter V provides recommendations on what to consider when interoperable medical devices develop.
- The chapter determines what information is expected to be the FDA in the observations.
Kapitel V: ‚‚ Considerations on design for interoperable medical devices’
The FDA requires that producers deal with the following aspects during the design of interoperable medical devices:
A) Purpose of the interface (electronic)
Manufacturers must determine in a purpose
- What data
- In which format,
- How (protocols, university/bidirectional etc.)
- Including which devices
- For the purposes (e.g. transmission alarm, control device)
- In which clinical context (e.g. when the patient ventilation)
it should be replaced.
The FDA not only requires the “Scope“(” Purpose “), but the Specify the interface.
B) proposed users
The FDA calls groups of users such as doctors, service staff, systems, IT and patient employees. These pursue different objectives when using the interface and have different knowledge.
Manufacturers must know these groups of users and take into account risk management.
C) risk management
Manufacturers are obliged to identify and master the risks due to the lack of interoperability. This includes risks due to incorrect data or controls, through interface malfunctions, through problems with IT security or the network (bandwidth, latency width, etc.).
Manufacturers must guarantee basic security and essential characteristics (see IEC 60601-1).
In addition, the FDA underlines that producers must analyze and master the risks on the complete life cycle of the product (i.e. not only in development, but also in the post-market phase).
D) verification and validation
The FDA also provides examples of what producers should consider when “test” (verification and validation):
- Returning corrupt data
- Safe operation also for data outside the value areas
- Transition to safe conditions
- Compliance with interoperability standards (if they have been defined)
- Authorization and authentication
- Usability
- Effects on the network and other devices and systems
- Manufacturers should run these tests in simulated use environments.
E) “labeling”
Labels, use and installation instructions and other “labeling” elements should also contribute to minimizing risks. The orientation document describes exactly what the labeling should contain in the part of the chapter VI D.
F) “consent standard”
The points of the FDA (recommended) to the “consent standard” without appointing them directly in the document. Rather, connect the list of these standards. Examples of these standards can be found listed below.
Use this chapter V to integrate your development plan or to check completeness!
Kapitel VI: ‚Recommendations for the contents of pre-market presentations’
In chapter VI, the FDA describes what information awaits you when sending “interoperable medical devices”. This information essentially corresponds to that mentioned in chapter V. Therefore, only some special characteristics of this chapter are mentioned here:
- The documentation should include: the purposes and technologies of the interface, standards used, type of data replaced, functional and non -functional requirements, possibly description of the API
- ISO 14971 and the needs of part 21 cf. 820.30 (G) must be observed.
- With critical applications such as infusion pumps, it is necessary to present all the specifications and results of the test. In the case of acritic, the summaries can be sufficient.
- The labeling must also meet the requirements of the 21 CFR part 801 and 809.
- The labeling (understanding for VA is probably the instructions for use) should describe the aspects mentioned in chapter V in subcontri A and B. to users should also have opportunities to verify the correct functioning of the interfaces.
The FDA has widely changed its QM system to ISO 13485. This also affects the requirements of part 21 cf. 820.30. Read more here.
3. Meet the requirements
A. Complete documents
The FDA orientation document “Interoperable medical devices” places the requirements for the development and documentation of medical devices. It is possible to implement these requirements by taking the following steps:
- Full purpose
Make sure that the purpose of your medical devices determines the points mentioned in this orientation document, such as
– clinical context
– devices to be binding
– users proposed
– Purpose of the data exchange - Specific of the requirements of the complement system/software software
Complete the required documents. Specify systems / software as a black box. Use the Blackbox model.
More information on this topic in the articles on the Blackbox model and on the requirements
Use consent standards where possible. You can read more on this below. - Complete instructions for use and installation
With these complete clauses your instructions for users. Use the chapter DI DI as a control list to ensure completeness. - Complete risks management watches
Now complete the watches for risk management. This affects the risk table and the risk management plan, which must also include the downstream phase. If necessary, adjust the post-market surveillance plan. It should oblige to search specifically and proactively risk information through interoperability problems. - Trial product
Carry out verification and validation activities. The specifications of the system or software requirements and the chapter there are input.
A great challenge will be to create system environments representative of the real environments.
B. contain “standard of consent”
The FDA has published a large list of Consenus standards as follows:
- ISO/IEEE 11073–10417 third edition 2017-04 COMPUTERS Healthcare – Communication for personal health devices – Part 10417: specialization of the device – Glucose metro.
- ISO/IEEE 11073–10418 First edition 2014-03–01 COMPUTERS – Communication for personal health devices – Part 10418: Specialization of the device: Normalized international monitor (INR) [including TECHNICAL CORRIGENDUM 1 (2016)].
As you can see from the date, this list has recently been updated.
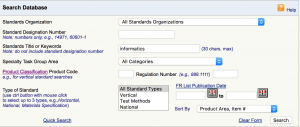
You can find these Consens standards on the FDA website, in which the “IT” key word is inserted in the “standard title or keywords” field:
Do not leave patient safety to chance
Sure it’s safe with a Pentes more from the Johner Institute!
4. Summary and conclusion
The FDA orientation document on “interoperable medical devices” is going on over the years. However, it is still useful. You can find more recent thoughts from the FDA on the interoperability on the FDA website “Interoperability of medical devices”.
It is misleading that the FDA only speaks of “legally non -binding” and “recommendation”. Finally, the American authority expects exactly this information mentioned in this document.
The orientation document does not call “consent standard”, but refers to the list of the published. This makes the document “waiting”. However, the list of consent standards is (too much) limited to medical (physical) devices and ISO/IEC 11071 standards. You would have liked more help for autonomous software.
Change the story
- 2025-03-09: updated document, structure of the modified chapter
- 2017-09-26: first version of the published document
Similar posts
Die FDA The importance of medical devices’ abuse recognized at the beginning and 2017 Dokument Guida ‚Interoperable medical devices’ Published. The American authority would like to take into account the fact that the interoperability of medical devices is important for health care on the one hand. On the other hand, problems with a lack of…