Way to avoid a PMA?

THE De novo procedurethe FDA also speaks of the “de novo program” and the “de novo submission process”, which is one of the approval procedures for medical devices in the USA.
Manufacturers can use this process – as the name “de novo” suggests – for new types of products.
This means that manufacturers would have to submit a de novo application for products for which there is no comparable predecessor device (“predicate device”) but which the FDA would likely classify as Class I or Class II.
1. Context and introduction
a) Statement of the problem
Manufacturers are not allowed to go through the 510(k) approval process for new products because there is no comparable legacy product. The FDA has not yet defined a class for these new products, which is why they would automatically fall into Class III (see FD&C Act, 513(f)(1)).
For new products or Class III products, manufacturers would actually have to go through a PMA approval process. However, this “pre-market approval” (PMA) is as expensive as it is laborious and time-consuming. Manufacturers of non-critical products would be overwhelmed.
b) Resolution approach
To limit these disproportionate efforts, the FDA initiated the trial de novo. It is a process by which the FDA requests new products
- class determines
- assign a new product code e
- defines the relevant “controls”. Examples of controls are special labeling requirements, design controls, or manufacturing controls, which are aspects of a quality management system. Here you will find an overview of the controls.
A product approved as “de novo” can itself serve as a “predicating device” for other similar products.
2. Developing the procedure de novo
a) Variants
There are two ways to start the procedure (see Fig. 1):
- Once again “classic”.
Manufacturers file a 510(k) application (PMN Premarket Notification). The FDA rejects this claim as the comparator product is not “substantially equivalent” (NSE). In the rejection, the FDA may express its assessment that the product to be approved is a “de novo candidate.” I could. Regardless of this assessment, producers can then submit a de novo application referring to the refusal. - „direct again“
Producers can now also make such a request directly. This is recommended if manufacturers believe there is no suitable predecessor product and the FDA will classify their product as Class I or II.
In the second case, manufacturers should also have one Pre-presentation request.
b) Examination by the authority and timing
Formal exam
First, the FDA reviews the application using an “Acceptance Checklist,” which can be found in Appendix A of the “Acceptance Review for De Novo Classification Applications” guidance document. The authority expects it within 15 days decides whether to accept the application or not.
First, it evaluates the application according to more formal decision-making criteria. This also includes if
- the producer paid the tariff,
- the product is actually a medical device,
- the question is directed to the correct FDA center and if
- this request can also be submitted for the product. For example, if there were a Predicate Device, this would not be the case.
Once the application clears this first hurdle, the FDA proceeds through the Appendix A checklist and reviews:
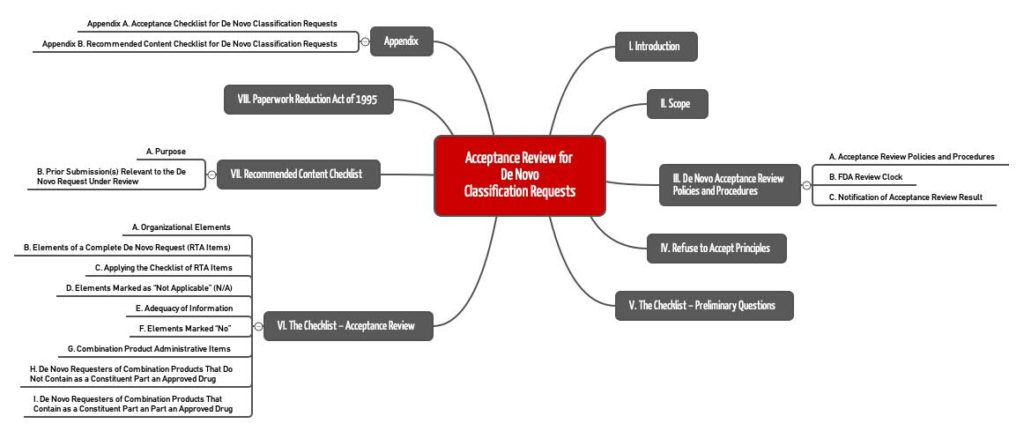
Examination of the content
For content review, FDA employees use the same guidance document, but the checklist in Appendix B. This checklist groups together questions on the following topics:
- Description of the device including accessories, labeling for patients and users, etc.
- Reason why the de novo procedure is requested.
- Mention of alternative processes, technologies, products
- Summary of preclinical data such as tests and clinical data from clinical studies
- List of standards adhered to (in particular “consensus standards”)
Between the acceptance of the application and the decision of the authority there should be no more than 60 Roofs die.
c) Documentation to be presented
The FDA requires similar information for both variants mentioned in point a). The FDA has already developed a framework for manufacturers to submit this information. For applications from October 1, 2025, submission is in eSTAR format. The FDA describes the structure and content of the guidance document Electronic Submission Template for Medical Device De Novo Requests. This requires, among other things:
- Administrative information (manufacturer/applicant, product name, type of presentation, etc.)
- Reference to any communications with the FDA and FDA history (e.g. IDE, pre-submissions) prior to submission
- Angewandte recognized consensus standards
- Name and detailed description of the product, including its intended purpose or indications for use
- Classification proposal with justification
- Brief description of benefits, risk analysis and description of risk minimization measures
- Justification of why the product is safe and achieves its intended purpose using scientific evidence (risk-benefit discussion). This typically requires clinical data on the product.
- For products that may fall into Class II, a suggestion on possible “special controls” to minimize risks
- “Labeling”, in particular instructions for use
- Information on preparation and its validation
- Information on sterilization and its validation
- Information on “shelf life”
- Biocompatibility assessment
- Information and special tests on software/firmware, cybersecurity and interoperability (if applicable)
- Test results (e.g. laboratory tests, software tests, animal experiments, clinical studies)
- References (e.g. to scientific articles)
- Responses to FDA Inquiries (“Requests for Additional Information”)
As described above, the FDA requires the application in eSTAR format with a specified chapter structure (see Fig. 3).
3. Regulatory context
The Food, Drug & Cosmetic Act describes the de novo process in section (2) of section 513(f). It also calls the procedure “Class III Automatic Designation Evaluation.”
This article clarifies that this procedure is only permitted for products that are not already classified.
4. Costs for a de novo procedure
A De Novo request is not cheap (see “Price List”): As of fiscal year 2025, the FDA requires more than $162,000. For small businesses, the FDA reduces the fee to just over $40,000.
5. Conclusion
The de novo process is the process of choosing new and less critical products. Even in this case, careful preparation is essential. Producers must clearly justify why
- because there is no comparable product,
- because they suggest a specific class and corresponding “controls”,
- what risks exist e
- how and why these are controlled so that the benefits outweigh the risks.
A clear purpose, a structured product description and test results are a prerequisite for this topic.
Support
The Johner Institute helps manufacturers quickly and without bureaucracy:
- Decide whether to submit a de novo application directly
- Check if the application is complete to avoid unnecessary waste of time or even rejection
- Decide whether a pre-sub or 510(k) filing is a better strategy
- Add missing information and improve applications
- Participate in pre-submission meetings
- Manage communications with the FDA
This allows manufacturers to minimize approval risks and therefore unnecessary costs and delays and bring products to the US market quickly and safely.
Are you waiting for your product to be approved in the United States? Then contact us now.
Change the history
- 2024-10-11: Revision due to eSTAR format required from 2025
Similar posts
THE De novo procedurethe FDA also speaks of the “de novo program” and the “de novo submission process”, which is one of the approval procedures for medical devices in the USA. Manufacturers can use this process – as the name “de novo” suggests – for new types of products. This means that manufacturers would have…