The guidelines for clinical exams

Die In ISO 14155: 2000 It is a standard with the title that has not been harmonized so far for the MDR “Clinical investigations on medical devices for human subjects – good clinical practice”. In German: “Clinical examination of medical devices – good clinical practice”.
He then describes the state of the art for producers of medical devices in the preparation, planning, implementation and evaluation of clinical tests and determines the responsibilities of the actors involved, in particular the sponsor so called.
This article helps with an Owning of ISO 14155 and with suggestions on the use of the standard to meet the regulatory requirements for clinical tests as quickly and without obstacles possible.
1. ISO 14155: an introduction
1.1 Context
The MDR has increased the requirements for the clinical data with which producers of medical devices must demonstrate the performance, safety and effectiveness of their products. Therefore, more producers are now required to collect these data in the context of clinical studies, in particular clinical exams. The MDR also puts extensive requirements for these clinical tests.
ISO 14155 should serve producers in the future for the MDR in the future.
1.2 ISO 14155 application area
On the one hand, the standard is applicable to clinical tests of medical devices that serve the goal of demonstrating the performance, safety and effectiveness of the products. On the other hand, the other “clinical studies after positioning on the market” (according to the formulation of the standard) also fall within the scope of ISO 14155. These are, for example, post-market clinical follow-up studies (PMCF).
1.3 objectives of the standard
The standard pursues several objectives:
- Protection of subjects (security, wellness, rights)
- Resilience of results
- Clearness on the responsibilities, in particular the sponsor (e.g. manufacturer), the main tester and potential other participants (for example CRO, hospital)
- Uniform understanding of all participants (sponsors, examiners, ethical commissions, authorities, appointed bodies)
1.4 Use for manufacturers
Therefore, the producers of medical devices benefit from the standard:
- Less regulatory uncertainty and discussion with the authorities and authorities appointed
- Consequently, faster and less complex “approval” of the products
- Time saving through concrete guidelines and examples, e.g. B. On the structure of documents such as the clinical test plan and the clinical tests report
2nd structure of ISO 14155: 2020
The standard includes over a hundred pages and consists of ten chapters and ten attachments (see fig. 2).
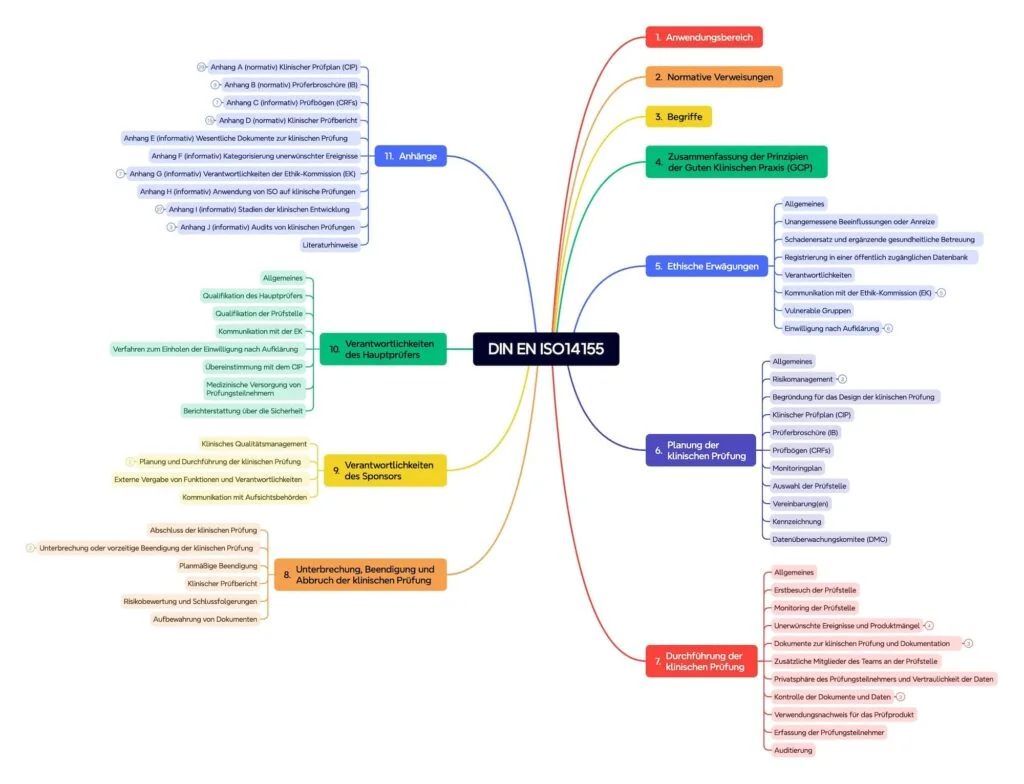
Chapter 4: good clinical practice
Chapter 4 summarizes the principles of good clinical practice (GCP).
Chapter 5: ethical considerations
Chapter 5 contains ethical considerations. Examine communication with the Ethics Commission, responsibilities, vulnerable groups, the declaration of consent and patients information.
Chapter 6: clinical examination planning
Chapter 6 describes the planning of the clinical test. This includes:
- Provisions on the test product
- Specific for the design of the clinical examination
- Specifications on the selection of the test center and on the essential documents of the clinical examination
These documents include:
- Clinical tests plan (CIP – Clinical investigation plan)
- Brochure test (IB – Cross of investigators)
- Archeses tests (CRFS – Forms of reporting of cases)
Annexes provide more information on creating these documents.
Chapter 7: Implementation of the clinical examination
Chapter 7 appoints the requirements for the implementation of the clinical test:
- Information on practical implementation
- Messages of events or product defects
- Management of documents and data
- Implementation of monitoring
- Confidentiality of data and identification of participants in the exam
Chapter 8: Interruption, resolution and demolition of the clinical examination
Here the requirements for the interruption, resolution and demolition of the clinical examination are included. These also concern the report on clinical tests, risk assessment and the conservation of documents.
Chapter 9 and 10: Responsibility
Chapter 9 and 10 regulate the responsibilities of the sponsor and the main auditor. The requirements for the sponsor are particularly relevant for producers of medical devices.
Regulatory and information attachments
The wide attachments of ISO 14155 are sometimes regulatory.
- Clinical tests (CIP) (regulatory): defined content of the CIP
- BROCHURE professor (IB) (regulatory): minimum requirements for the IB content
- Archeses tests (CRFS) (information): suggestions for the creation of CRFS
- Report on clinical tests (regulatory): defined content of the clinical test relationship at the end of the clinical examination
- Essential documents for clinical tests (information): documents that must be available before, during and after clinical examination at the sponsor or the test center
- Categorization of unwanted events (information): panoramic table and graphics for orientation
- Responsibility of the Ethics Commission (EK) (information): Guide to good practices in ethical commissions
- Application of ISO 14971 to clinical examinations (information): graphics to face risks during a clinical examination
- Clinical development stadiums (information): overview of the various types of accompanying the development of clinical tests of the medical device and the applicability of ISO 14155 on the various types
- Audit of clinical exams (information): suggestions on how sponsor can demonstrate that clinical examination takes place in harmony with GCP
3. ISO 14155 important requirements
3.1 ethical considerations
Clinical tests must be carried out in accordance with ethical principles. ISO 14155 must always be used with the current version of the Helsinki declaration, in which the ethical principles for the implementation of clinical exams are defined.
The foreseeable risks must be evaluated with respect to the expected benefit.
The rights, safety and well -being of the participants have a priority before scientific interests.
3.2 unconditional scientific rigorosity
The exam must follow clear and understandable statistical requirements that bring well -founded results with high tests.
Preclinical and clinical information must sufficiently support the test.
3.3 Complete documentation
In -depth and understandable documentation of all phases of the clinical test is required.
The implementation must be carried out in accordance with the documents approved by the Ethical Commission and by the Authority (UA test plan, clinical examinator manual, patient information).
3.4 Clear responsibilities
Clear definitions of the roles and responsibilities of all the people involved are essential and must be determined in writing.
4. Stop blocks that must be avoided
4.1 Conditions discovered at the manufacturer
The hypothesis that the manufacturer can easily take on the role of the sponsor in the clinical test can often be taken.
The sponsorship within a clinical study is associated with some specifications, including the integration of the management of clinical quality in the manufacturer’s existing QMs.
So keep in mind that you can start the exam only if as a sponsor meets the requirements of the standard and the MDR.
4.2 ISO 14155 applicability error
“We only do a PMCF study, the product is already certified, so the norm does not apply.”
This is a misunderstanding.
The standard applies not only For the “admission studies” that are conducted to obtain the EC. It must also be used in parts for studies on the clarification of scientific questions (article 82 according to MDR) or in PMCF studies (Article 74 studies). This is explained in Appendix I.7 of the standard.
4.3 Missing documentation
As a rule, the summary of the results of the study is not enough to demonstrate clinical tests.
Manufacturers must evaluate the clinical data of their product by studies as part of the clinical evaluation. If you perform a study with your product, you expect you to present the study of the study to demonstrate clinical tests and to be able to demonstrate the compliance of studies with ISO 14155. This is controlled by the body indicated as part of the “evaluation of the clinical evaluation” (section and of the MDCG 2020-13).
In the worst case, doubts could arise on the quality of the data, since a manufacturer cannot demonstrate without the test plan and the other documents that the study, e.g. B. was carried out according to ISO 14155. This would mean that the data should be excluded if necessary.
4.4 Exclusive focus on ISO 14155
It is a widespread error that pure compliance with ISO 14155 automatically meets all national requirements. On the other hand, it is correct that national requirements, in particular outside the EU, must be taken into consideration.
5. Versions of ISO 14155
5.1 Differences between the 2012 and 2020 versions
ISO 14155: 2020-01 (or DIN EN ISO 14155: 2021-05) has implemented several noteworthy changes compared to the preliminary form of 2012:
- The intertwining with the ICH-GCP guideline was highlighted and a summary of the GCP principles was integrated into the standard in section 4.
- Risk management was integrated in the entire clinical test process.
- Appendix I was introduced, particularly interesting for producers, which explains the applicability of the standard to the different levels of clinical development of the medical device.
- A request was added to record the clinical examination in a database accessible to the public (new in the 2020/2021 version).
- Clinical quality management (section 9.1) and risk -based monitoring (section 6.7) have been integrated.
- The adaptation to the international data protection requirements was taken into consideration in the current version.
5.2 Outlook for the next version
A new version of ISO 14155 (DIN EN ISO 14155/A11: 2024) is currently being developed. The publication time of the final standard is still unknown (in June 2025).
The revision also aims to harmonize clinical tests with the EU requirements of the MDR. It should be valid without a transition period. (Source)
6. Conclusion and summary
ISO 14155 – regardless of its harmonization – is the Gold Standard for the preparation, planning, implementation and evaluation of clinical studies on medical devices. It is not only applicable to clinical exams and should also be followed in studies outside Europe.
The standard is sufficiently precise and complete and therefore to the most beneficial producers of another regulatory burden.
The “clinical experts” of the Johner Institute will help you find clinical data and decide if a clinical study is still necessary.
In this case, we will support you in your complete clinical examination or in the PMCF study: from planning to application to implementation.
- Study design: we help you design and vote.
- Planning of the statistical case number: optimal planning for valid results
- Cro-Selection: we find the right research organization on the contract (CRO).
- Presentation to the authorities: creation of regulatory documents
- Implementation and evaluation: always by your side while you work with the CRO in the clinical field
Here you will find an overview of the support of the Johner Institute for reviews and clinical exams.
Similar posts
Automotive
Berita Olahraga
News
Berita Terkini
Berita Terbaru
Berita Teknologi
Seputar Teknologi
Drama Korea
Resep Masakan
Pendidikan
Berita Terbaru
Berita Terbaru
Berita Terbaru
Die In ISO 14155: 2000 It is a standard with the title that has not been harmonized so far for the MDR “Clinical investigations on medical devices for human subjects – good clinical practice”. In German: “Clinical examination of medical devices – good clinical practice”. He then describes the state of the art for producers…