Everything you need to know

The European one Medical Devices Regulation MDR (EU Regulation on Medical Devices) must be taken into consideration by manufacturers intending to market medical devices in the EU.
This Regulation (EU) 2017/745 on medical devicesthe official title, also imposes requirements on notified bodies, traders, importers and healthcare facilities such as hospitals.
… for beginners
If you are brand new, download this free starter kit below. It offers you an overview of the legislation, shows you the steps for “approval” of your medical device and contains the MDR checklist in PDF and DOCX format for download.
… for advanced users
Learn more about the details with this page and the specialist articles linked to it.
Use that Consolidated version of the MDR in German or English. This summarizes all changes to the MDR, including the transition periods extended to March 2023. Internal links make it easy to navigate the regulation, which is over 170 pages long.
… for IVDR manufacturers
Manufacturers of in vitro diagnostic instruments should read the specialist article on IVDR.
… for importers, retailers and operators
Watch the following video and then read the specialist articles linked in chapter 1.b).
1. Overview of MDR requirements
1.1 Requirements for manufacturers and their medical devices
The requirements imposed on manufacturers by the Medical Devices Regulation are numerous.
1.1.1 Requirements transversal to the product
| MDR requirement and links to specialist articles |
Explanation |
| Quality management system | All manufacturers need a QM system, among other things, for the development, production and monitoring of products on the market. With the exception of Class I products, certification is usually required. |
| Risk management system | Risk management must ensure throughout the product life cycle that the benefits are acceptable given the risks. |
| Verantwortliche Person (“Person responsible for regulatory compliance”) | Manufacturers are required to employ a person responsible for regulatory compliance and e.g. B. ensures that technical documentation is created in accordance with the company’s own QM system. |
1.1.2 Product-specific requirements
| MDR requirement and links to specialist articles |
Explanation |
| Product classification | The manufacturer must determine the risk class for each product. |
| Fundamental safety and performance requirements and technical documentation | The technical documentation must meet the requirements of Annex II and provide all evidence of compliance with the essential requirements of Annex I. For this proof, manufacturers are required to use harmonized standards and common specifications. |
| Clinical evaluation | As part of the clinical evaluation, the manufacturer must continuously verify whether the safety, performance and benefits of the products are met. If clinical data are not sufficient, a clinical trial is necessary. |
| Unique Device Identification (UDI) | All medical devices must receive a unique identification, the UDI. The products must therefore be registered in EUDAMED. |
| Labeling | The MDR precisely specifies requirements for instructions for use, other accompanying materials and other labeling such as printouts and packaging. |
| Conformity assessment | Depending on the risk class of the product, the manufacturer may undergo a conformity assessment procedure. To this end it must involve a notified body, with the exception of class I products. If successful, it will have to complete a declaration of conformity and affix the CE marking. |
| Post-market surveillance and surveillance | Manufacturers are obliged to monitor their medical devices placed on the market throughout their useful life, to continuously collect data and, if necessary, react to them. If there are risks it is necessary to inform the authorities. |
1.2 Requirements for other actors (retailers, importers, etc.)
Not only medical device manufacturers have to comply with the Medical Device Regulation, but also other actors:
2. Structure and structure of the MDR
The Medical Devices Regulation is made up of 123 articles, divided into 10 chapters. It also has 17 appendages.
2.1 The chapters
The Medical Devices Regulation (MDR) is completely restructured compared to the old Medical Devices Directive (MDD):
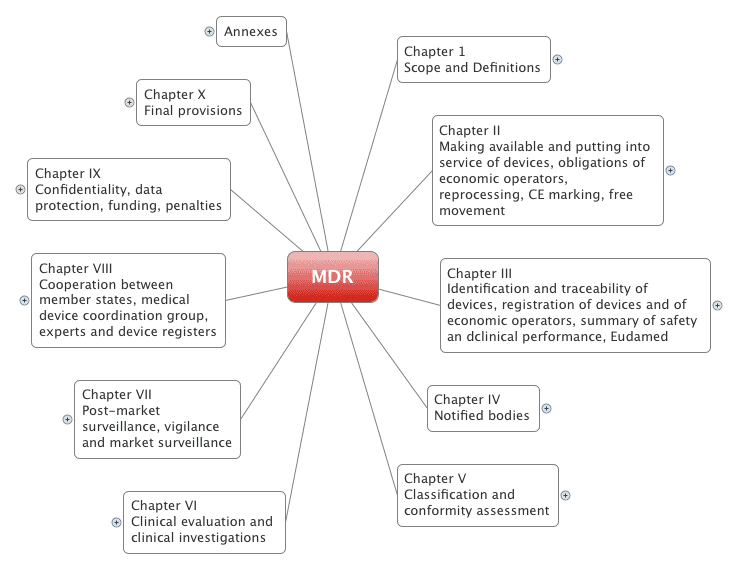
It provides a quick overview of the MDR, which contains more than 170 pages.
2.2 The appendices
The MDR has 17 appendices.
3. Further articles and specialist links
3.1 Other specialist articles
The EU has postponed the transition periods for moving to the MDR several times. The rules are so complex that a specialist article on transition periods will help.
The Medical Device Coordination Group (MDCG) has published further explanations and requirements.
There are different requirements for products without a medical purpose (‘Annex XVI products’).
3.2 Further connections
- MDR (original text)
- To be corrected
- Various
4. Basic information
4.1 How community regulations were born
It is often said that the breast implant scandal was the trigger for the revision of the Medical Devices Act. But most actors now deny it. It is therefore largely unclear who initiated the new regulation and for what reason.
4.2 Difference between community regulations and community directives
The “old” EU medical devicesGuidelines Like all EU directives, they had to be translated into national laws and regulations to gain legal force. In Germany these were the Medical Device Act (MPG) and regulations such as the Medical Device Operators Ordinance (still valid) and the Medical Device Safety Plan Ordinance (now invalid).
Dying EU-Regulations (here: MDR, IVDR) have a direct legal character. National laws simply supplement these rules, such as the MPDG in Germany, with criminal provisions and the determination of the competent authorities.
4.3 Criticisms of the Medical Devices Regulation
Criticism of EU legislation on medical devices is enormous. The efforts and costs for producers have multiplied. The duration of the “approval process” also applies.
As a result, competitiveness, innovation and supply of medical devices are hampered.
These consequences were already feared in 2020.

By uploading the video you accept YouTube’s privacy policy.
Find out more
Upload videos
==
For future regulation it is important to understand the system: a task of regulatory science.
5. Conclusion and summary
The MDR Medical Devices Regulation is a very broad piece of legislation that poses great challenges to all actors (medical device manufacturers, notified bodies, importers, retailers, hospitals). There is no evidence that MDR improves the safety, performance and effectiveness of medical devices. However, it is clear that the supply of medical devices is suffering.
Manufacturers have no choice but to intensively engage with this body of law and meet its requirements. These increased requirements are not limited to the “approval” (pre-market phase), but also explicitly influence the post-market phase (post-market surveillance, vigilance).
Help with MDR implementation
Free offers
Still have questions about MDR and its implementation? You can get answers in our free micro-consultation.
Download this free starter kit which offers an overview of the regulatory landscape and contains the MDR checklist in PDF and DOCX format.
Video and e-learning
Video training courses in Auditgarant show you step by step how to create your technical documentation and QM system in a lean, fast and MDR compliant way. Over 100 templates and sample documents are available for download. This way you will create the conditions for your products to be approved and placed on the market quickly and safely.
Product testing
Johner Institute experts will help you test your products:
Advise
Leverage the expertise of the Johner Institute’s regulatory affairs experts to…
Contact us now so we can clarify together how you can quickly and easily satisfy the regulatory requirements of the MDR and bring your products to the market in complete safety.
make contact
Change history (as of May 2021)
- 08-27-2024: In chapter 3.2 the links to the corrections have been corrected and the links have been grouped hierarchically
- 2024-01-18: Chapter numbering has been changed
- 2023-04-18: Article completely rewritten
- 2021-10-10: Added paragraph 7.e) (Consequences have not been considered and are not understood)
- 2021-07-26: Added link to new corrigendum
- 2021-05-24: New DoC post linked
Similar posts
The European one Medical Devices Regulation MDR (EU Regulation on Medical Devices) must be taken into consideration by manufacturers intending to market medical devices in the EU. This Regulation (EU) 2017/745 on medical devicesthe official title, also imposes requirements on notified bodies, traders, importers and healthcare facilities such as hospitals. … for beginners If you…